
In this Formaspace laboratory report, we take a look behind the headlines at the machine learning algorithms that have helped the AlphaFold project predict more than 214 million protein structures.
Why Understanding the 3D Structure of Proteins is Difficult yet Vital for Biological Research
Understanding the 3D structures of a protein can be enormously useful in uncovering the function and evolution of specific metabolic processes, such as identifying the cause (and hopefully cure) of a disease or evaluating whether an animal protein can be used as a human disease research model.
Yet visualizing the complex 3D structure of proteins accurately has remained a challenge, requiring time-consuming, painstaking direct observations using X-ray crystallography or (increasingly) cryo-EM techniques to create density maps of the protein structure in 3D space.
Researchers have long been looking for a shortcut, asking whether it would be possible to use our knowledge of the sequence of amino acids found in each protein to predict what its 3D structure would look like.

Why are Protein Structures so Complicated?
To borrow the words of Winston Churchill (speaking about Russia in 1939):
“It’s a riddle, wrapped in a mystery, inside an enigma.”
Calculating 3D protein structures is hard. To illustrate this, here is a breakdown of the first three ways they can fold in 3D space:
1) Primary Protein Structure
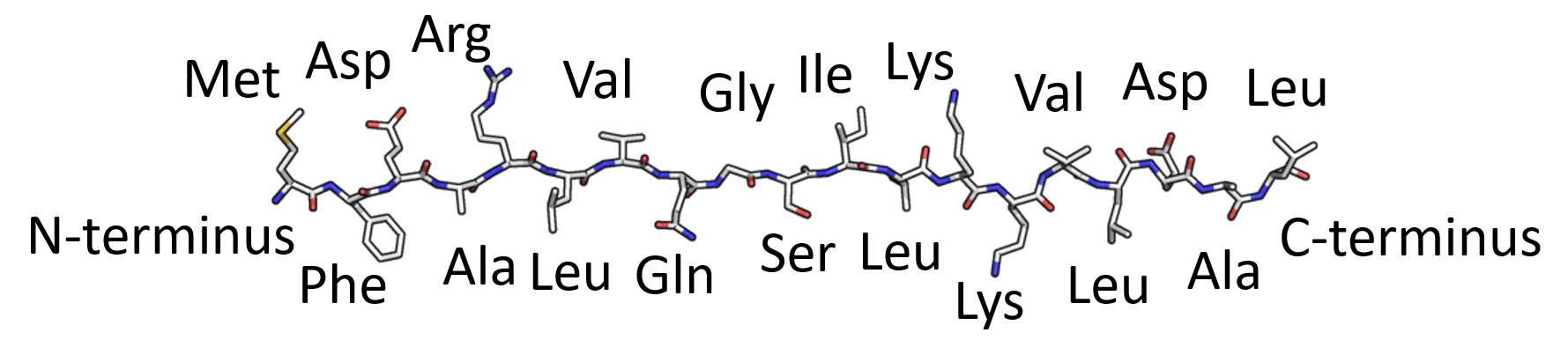
2) Secondary Protein Structure

3) Tertiary Protein Structure

CASP, the Bi-Annual Scientific Challenge for Predicting 3D Protein Structures
As you can imagine, it’s challenging to derive the 3D version of a protein if all you have to work with is the original sequence of amino acids in the chain.
But where there is a daunting scientific challenge, there is often a contest designed to spur innovation.
In 1994, Professor John Moult, Institute for Bioscience and Biotechnology Research at the University of Maryland, co-founded CASP, the Critical Assessment of Techniques for Protein Structure Prediction.
Every two years, CASP holds an international protein folding prediction contest that has drawn participation from over 100 top research groups from around the world.

What’s Behind AlphaFold’s Prowess in Predicting 3D protein structures?
The AlphaFold project is one of several emerging AI-based tools that can predict the complex 3D structure of proteins.
AlphaFold is a project of DeepMind; the London-based company co-founded in 2010 by Demis Hassabis, which was subsequently acquired by Google’s parent company, Alphabet, in 2014.
The DeepMind team first achieved widespread fame when its artificial intelligence-based AlphaGo program beat world champions playing the world’s oldest board game, Go.
In 2016, the organization turned its expertise to the problem of predicting protein folding.
They entered the CASP competition for the first time in 2018 (at CASP 12), where the AlphaFold team took top honors in predicting moderately difficult protein targets.
The AlphaFold team returned in 2020, where they once again swept the board at CASP 12, achieving a near 90% accuracy score in predicting 3D protein structures.
In July of this year, Deep Mind announced they had identified 214 million 3D protein structure predictions, making them freely available for public use.
It’s already having a dramatic effect on medical research.
For example, at the University of Oxford, AlphaFold protein predictions have sped up laboratory research on the parasites that cause malaria, helping lab researchers quickly identify target sites where antibodies could attach to proteins and block disease transmission.
How does AlphaFold Make 3D Protein Structure Predictions?
The secret behind AlphaFold’s dramatic achievements is machine learning, which is proving to be a powerful tool for solving problems that have a defined set of boundary conditions that could result in several possible outcomes.
Examples of where machine learning has met with success include recognizing text on images or video, rapid two-way translation of spoken or written human languages, interpreting medical scan imagery for signs of disease, creating sophisticated artwork based on keywords, and even powering autonomous self-driving cars (this last point, of course, remains a work-in-progress).
Training the Machine Learning Data in AlphaFold, a Brief Overview
Let’s look at a schematic diagram from Kathryn Tunyasuvunakool, Research Scientist at DeepMind. (For easier identification, we have added labels A through H.)
![]()
· Input Sequence A
This is the known amino acid sequence of the protein that we are trying to predict its corresponding 3D structure.
· Training Sets B, C, and D
Many machine-learning programs rely on training sets to “teach” the algorithm about the real-world problems it seeks to solve.
In the case of AlphaFold (and machine-learning operations from other competing research teams), the training set information is sourced from the international Protein Data Bank (PDB), which catalogs protein structures verified by X-ray crystallography, NMR spectroscopy, Cryo-EM, or other advanced methods.
This PDB information is reformulated into three training sets to be used by the machine-learning transformer networks:
- B) Multiple Sequence Alignments (MSA), which are short amino acid chain sequences shared (e.g. “aligned”) among related proteins, such as chymotrypsin (an enzyme that breaks down other proteins during digestion) which is found in humans and other species. Identifying related proteins with significant alignments can often help “guide” the machine learning algorithm toward a more accurate solution.
- C) Analysis of amino acid pairs (also known as residue pairs) can help predict the structure of proteins, as some amino acid residue pair combinations are statistically more likely to maintain close contact with each other, thus providing a useful clue for the final 3D structure.
- D) (Optionally) Templates. These are defined 3D protein structures which help inform the machine learning transformer networks. Ideally, these templates have been validated through direct observation (via X-ray or Cryo-EM), but even theoretical structures (even those predicted by AlphaFold itself) can be used to improve results.
· Machine Learning Transformer Networks
If you are familiar with machine learning techniques, the transformer networks used in AlphaFold will be familiar – at least at a very high level.
If not, Computerphile offers a short introduction to convolutional neural networks.
In our diagram above, the MSA representations (B) and Amino Acid Residue Pair representations (C) are evaluated 48 times in the “Evoformer” (E) which seeks to predict likely relationships.
The next transformation takes place in the Structure Module (F), which uses any available Templates (D) to evaluate the information 8 times to predict the likely rotations and position locations for each amino acid residue segment. You can think of this as a first draft of the 3D structure.
This is refined further in three additional steps:
- The first refinement is to consider the dihedral angles between the amino acid side chains and the main protein chain in 3D space. These dihedral angles are not arbitrary, they tend to follow certain stereochemical rules, and some combinations don’t occur naturally, which can help AlphaFold avoid making incorrect protein structure predictions.
- The second refinement is to perform a “relaxation” or energy minimization process, which allows the model to settle into a more stable, lower energy state. During this process, any remaining stereochemical violations or unrealistic bond lengths or angles are addressed.
- Finally, the proposed 3D protein structure (G) can be “recycled” through the system multiple times (3 by default) to iteratively “train” the system (H) – in practice, this has been found to improve and refine the final output (G) significantly.

How You Can Access AlphaFold Protein Structure Predictions
AlphaFold 3D protein structure predictions are free to access.
One of the easiest ways to access them is via the UniProt website, which is commonly used by biology researchers, especially those in the field of bioinformatics, who seek to understand the structure of proteins.
If you haven’t used the UniProt website, take the tour.
Why it’s Important to Treat AlphaFold Protein Structures as Hypotheses unless Independently Verified
We’ve been careful to refer to the output of AlphaFold as 3D protein structure predictions.
As noted Los Alamos National Laboratory researcher Tom Terwilliger points out in his recent paper, Improved AlphaFold modeling with implicit experimental information, it’s important to recognize that AlphaFold predictions are a great starting point, they are hypotheses and should be treated with care.
In his paper and an online video, Terwilliger outlines how researchers use direct observation methods (such as X-ray crystallography or Cryo-EM) to refine individual AlphaFold protein model predictions through iterative training.
What’s Next on the Horizon?
The next challenge facing systems such as AlphaFold is to do a better job predicting larger protein structures, such as Quaternary protein structures (also known as multimers).
· Quaternary Protein Structure (Multimer)

Due to limitations in current technology, large protein structures often exceed the available memory resources necessary to run these larger models through machine learning transformer networks (even for well-resourced computers from DeepMind.)
However, researchers are rapidly making advances in creating alternate workflows to make useful predictions for multimer proteins.
Meanwhile, there is a wish list for future developments.
Researchers would like to have access to tools that can predict the structures of diseased or otherwise compromised proteins.
Understandably, they would also like to increase the scope of predicted structures to include DNA, RNA, carbohydrates, ligands, ions, and covalent modifications, as well as interactions with water.
As with all branches of science, there is always more to discover.
We can imagine that this complicated array of moving things which constitutes “the world” is something like a great chess game being played by the gods, and we are observers of the game. We do not know what the rules of the game are; all we are allowed to do is to watch the playing. Of course, if we watch long enough, we may eventually catch on to a few of the rules… If we know the rules, we consider that we “understand” the world.
— Nobel Prize Physicist Richard P. Feynman (1918-1988)

Formaspace is Your Partner for Bioinformatics and Laboratory Research
If you can imagine it, we can build it – here at our factory headquarters in Austin, Texas.
Talk to your Formaspace Design Consultant today and find out how we can work together to make your next laboratory project or remodel a complete success.









